IRG-II: Catalytic Materials for CO2 Reduction
The development of stable, inexpensive catalysts that lower the activation energy for reactions and reduce the production of undesirable side products is one of the most challenging aspects of materials science, and is vital to the solution of many problems facing humanity. Understanding basic chemical principles to the degree that a particular catalyst can be efficiently designed for a given purpose remains beyond the grasp of today’s chemistry. Nowhere is this more apparent than in the realm of energy conversion. For example, photovoltaic solar cells can produce electricity using sunlight (IRG I), but electricity can be used to fill all of society’s energy needs only if it can be used to produce fuels such as reduced carbon materials or hydrogen efficiently. Fuel cells (IRG III) can convert such fuels to electricity only if suitable catalysts for both electrode reactions can be identified.
How can effective catalysts be designed? There is an existing paradigm – biology. The biological world functions through the production of highly specific, highly effective enzymic catalysts. Enzymes can be found that carry out a huge host of chemical transformations, including many of those of importance for energy conversion – water oxidation, carbon dioxide reduction, oxidation of reduced carbon compounds, hydrogen gas production and oxidation. Perhaps technologically useful catalysts can be found by emulating and adapting the mechanism that nature uses to develop biological catalysts. In this IRG, we will use such an approach to synthesize and identify new catalysts for the reduction of carbon dioxide to useful fuels such as methanol.
Enzymes are made up of proteins, and often include organic or inorganic cofactor molecules. Biology basically takes a simple reaction that can perform the desired chemistry, but is neither rapid enough nor specific enough for the required purpose, and tunes it using the protein environment in order to lower the activation energy and exclude side reactions. This tuning is achieved through evolution of the peptide framework and the cofactor via genetic changes that occur in organisms over long periods. In this project, we will use a new chemical evolutionary approach to find peptide-based catalysts, and will couple this with extensive theoretical modeling to limit the structure space which must be searched. The approach aims to capture the best aspects of both chemical evolution and rational design.
 Carbon dioxide can be reduced on bare metal or glassy carbon electrodes, but only at very negative potentials (-2.2 V vs SCE) due to extremely high overpotentials. This is because the first steps in the reduction: Carbon dioxide can be reduced on bare metal or glassy carbon electrodes, but only at very negative potentials (-2.2 V vs SCE) due to extremely high overpotentials. This is because the first steps in the reduction:
CO2 + 2e- ® CO22-
require two electrons, resulting in substantial uncompensated charge on the product. Catalists substantially reduce the activation energy associated with the reaction, increasing speed and efficiency because the two reduction steps occur within the coordination sphere of the metal, thereby stabilizing the intermediates and products. Using such complexes, the overpotentials for electrochemical CO2 reduction in solution have been reduced by as much as 0.5 V. However, the energy efficiency of CO2 reduction using these catalysts, while improved, is not as high as it needs to be for practical applications. Also, in their current form, these catalysts are easily inactivated by extraneous materials or side reactions of the reduction process, and give a heterogeneous set of products (such as methanol, methane, carbon monoxide and formate), making them impractical for broad use.
We propose to modify such metal porphyrin catalysts, improving their catalytic efficiency, chemical robustness, and product specificity. The chemical environment of the metal center will be adjusted to lower the overpotential for CO2 reduction, and the reaction site will be tuned to reduce side reactions and to produce products of interest. Our initial target product will be methanol, which can be used in fuel cells or burned directly. During this tuning process it will be imporant to maintain simplicity of fabrication. Such simplicity is vital if the project is to succeed within a reasonable time frame, and if the final products are to be technologically useful.
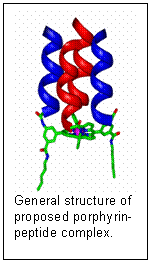 Our approach is to derivatize the aryl groups attached to the porphyrin macrocycle with peptides, that will impart efficiency, specificity, protection from side reactions, etc., much as the peptides in enzymes tune the chemistry of their active sites. Heme-containing proteins, for example, carry out a wide variety of small-molecule binding and redox functions, depending upon the peptide environment around the porphyrin. We will build cobalt or iron porphyrin/peptide complexes in which a four-helix peptide bundle is covalently attached to the porphyrin, forming a catalytic pocket reminiscent of an enzyme. Such a design provides many degrees of freedom for developing molecular recognition properties that can mediate substrate access, catalytic function and reaction specificity. The peptides will be formed from the 20 amino acids found in enzymes, but may also include non-natural amino acids with special functionality. Our approach is to derivatize the aryl groups attached to the porphyrin macrocycle with peptides, that will impart efficiency, specificity, protection from side reactions, etc., much as the peptides in enzymes tune the chemistry of their active sites. Heme-containing proteins, for example, carry out a wide variety of small-molecule binding and redox functions, depending upon the peptide environment around the porphyrin. We will build cobalt or iron porphyrin/peptide complexes in which a four-helix peptide bundle is covalently attached to the porphyrin, forming a catalytic pocket reminiscent of an enzyme. Such a design provides many degrees of freedom for developing molecular recognition properties that can mediate substrate access, catalytic function and reaction specificity. The peptides will be formed from the 20 amino acids found in enzymes, but may also include non-natural amino acids with special functionality.
The chemical diversity possible even with four short peptides and the twenty usual amino acids is still too large to explore efficiently, even using combinatorial techniques at the scale mentioned above. For this reason, we will combine this high-throughput synthetic approach with a novel high-throughput computational modeling method that will allow us to both design libraries of potential catalysts and correlate calculated structural parameters with measured function. Traditional molecular dynamics simulations of peptides or peptide complexes are currently too slow to consider using for the kinds of screening calculations we would like to do here. Instead, we will use an exciting new geometrical simulation technique (FRODA, developed by a member of the research team) that can explore local peptide conformations about a thousand times faster than can be done with conventional molecular dynamics simulations, and is therefore well suited to high throughput virtual screening coupled with feedback from experimental results.
The work will require expertise in peptide design, porphyrin chemistry, photochemistry, laser scanning, electrochemistry and large scale calculations. Woodbury’s expertise is in photochemistry and photobiology, and his laboratory already has experience in both light-directed and electrochemically-directed synthesis of catalyst arrays. Gust is a leading expert in the synthesis, photochemistry and electrochemistry of porphyrins and will guide the design, synthesis and analysis of the porphyrin macrocycles and their coupling to peptide chains and to electrically active surfaces. Ghirlanda is an expert in peptide structure and function and will design candidates for the peptide binding pocket that defines the catalytic site. Finally, Thorpe is a theoretical physicist who has developed new approaches to the modeling of biological macromolecules. His group will perform the high-throughput structural modeling.This specific goal of this project is the discovery of useful new catalysts for conversion of CO2 to a reduced carbon fuel such as methanol. More importantly, however, it will serve as a vehicle for developing a general new approach to designing bioinspired catalysts that couples our knowledge of enzyme function, modern computational theory, and random chemical evolution methods. This information will contribute to the future design of catalysts for many important processes, including catalysts for the redox reactions of fuel cells (IRG III).
|